
TESTES DE TRIAGEM NEONATAL
O termo triagem, do francês triage, significa seleção, separação de um grupo, e define, em Saúde Pública, a ação primária dos programas de detecção – testes aplicados numa população – de um grupo de indivíduos com maior probabilidade de apresentarem determinadas patologias.
Desde a década de 60, a Organização Mundial da Saúde (OMS) preconiza a importância da realização dos programas populacionais de Triagem Neonatal, especialmente nos países em desenvolvimento, além de criar critérios para a realização dos mesmos.
No Brasil, desde 2001 foi implementado o Programa Nacional de Triagem Neonatal. Para que um defeito metabólico seja considerado importante para um procedimento de triagem, certos critérios devem ser observados, como orientado por documento da OMS:
- não apresentar características clínicas precoces
- permitir a realização de um teste de identificação com especificidade e sensibilidade altas
- ser um programa economicamente viável
- estar associado a uma doença cujos sintomas clínicos possam ser reduzidos ou eliminados através de tratamento
- ter estabelecido um programa de acompanhamento clínico com disponibilização dos quesitos mínimos necessários ao sucesso do tratamento.
Inicialmente, foi implementado o Teste do Pezinho. Trata-se de ação preventiva que visa facilitar o diagnóstico precoce de certas doenças, reduzindo ou eliminando o risco de sequelas associadas ao atraso no reconhecimento e tratamento dessas condições. Toda criança nascida em território nacional tem o direito à triagem neonatal (Teste do Pezinho).
Mas, para que este alcance o seu objetivo de detectar algumas doenças que podem causar sequelas graves ao desenvolvimento e crescimento, o teste deve ser feito no momento e da forma adequados.

O momento para a coleta, preferencialmente, não deve ser inferior a 48 horas de alimentação amamentação e nunca superior a 30 dias, sendo o ideal entre o 3º e o 5º dia de vida.
É muito importante respeitar o tempo de realização. O teste não deve ser feito antes das 48h de vida, pois pode sofrer influência das alterações hormonais e metabólicas que naturalmente acontecem com a transição da vida fetal para a vida pós-natal, e que demoram até dois a três dias para atingir o equilíbrio.
Já a coleta tardia pode impedir que a suspeita e o diagnóstico sejam feitos no tempo correto e, consequentemente, atrasar as intervenções e tratamento específicos, fazendo com que a doença provoque consequências que podem ser irreversíveis, prejudicando o crescimento e desenvolvimento.
O exame é feito através de uma pequena picada no calcanhar do bebê. O furo é praticamente indolor e o sangue é coletado rapidamente. O calcanhar é o local escolhido por ser uma região com muitos vasos sanguíneos.

As gestantes devem ser orientadas, ao final de sua gestação, sobre a importância do teste do pezinho e procurar um posto de coleta ou um laboratório indicado pelo pediatra dentro deste prazo.
Os laboratórios privados realizam testes para outras doenças, cabendo ao pediatra selecionar as que são de interesse.

Nos casos com resultados de triagem alterados, o laboratório central deve acionar o posto de coleta para que entre em contato com a família e trazer a criança para a realização de exames confirmatórios.
Esse processo precisa ser ágil e eficaz para que a terapêutica precoce adequada possa ser instituída.
Importante: o Teste do Pezinho é apenas um teste de triagem. Um resultado alterado não implica em diagnóstico definitivo de qualquer uma das doenças, necessitando, de exames confirmatórios.
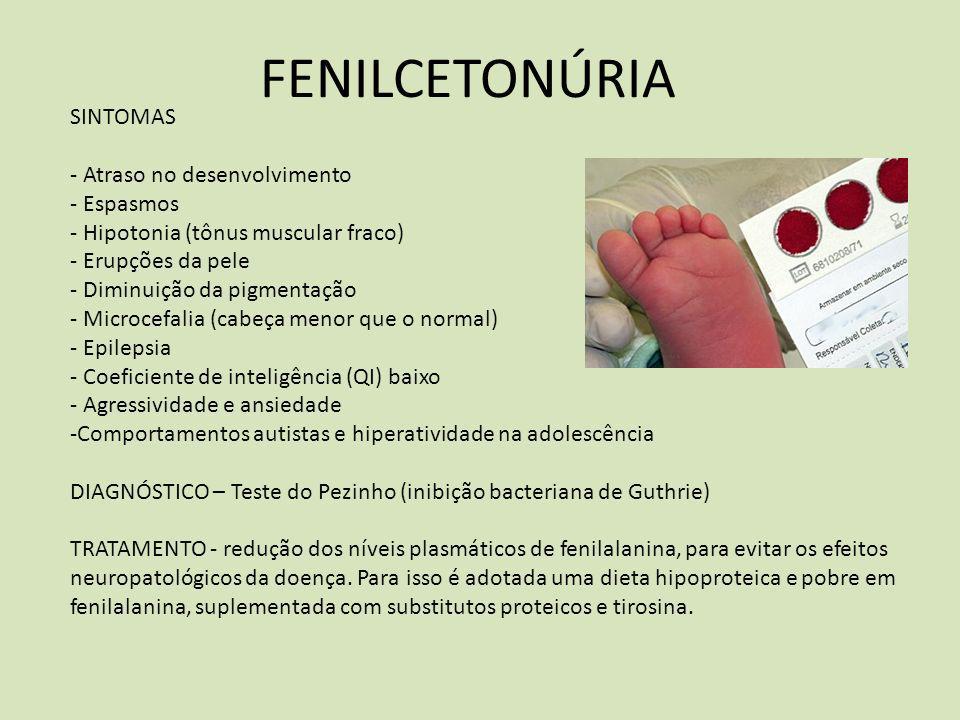
Fenilcetonúria
Doença genética em que a criança não é capaz de metabolizar o aminoácido fenilalanina existente em todas as formas de proteína da nossa alimentação (carne, leite, ovos etc.).
 Com isso, a fenilalanina se acumula no sangue e em todos os tecidos. Este excesso provoca lesões graves e irreversíveis no sistema nervoso central (inclusive o retardo mental) e o seu tratamento precoce pode prevenir essas sequelas.
Com isso, a fenilalanina se acumula no sangue e em todos os tecidos. Este excesso provoca lesões graves e irreversíveis no sistema nervoso central (inclusive o retardo mental) e o seu tratamento precoce pode prevenir essas sequelas.
Hipotireoidismo congênito
Distúrbio causado pela produção deficiente de hormônios da tireoide, geralmente devido a um defeito na formação da glândula, ou a um problema bioquímico que ocorre na síntese dos hormônios tireoidianos.
Os hormônios tireoidianos são fundamentais para o adequado desenvolvimento do sistema nervoso. A sua deficiência pode provocar lesão grave e irreversível, levando ao retardo mental grave. Se instituído bem cedo, o tratamento é eficaz e pode evitar sequelas.

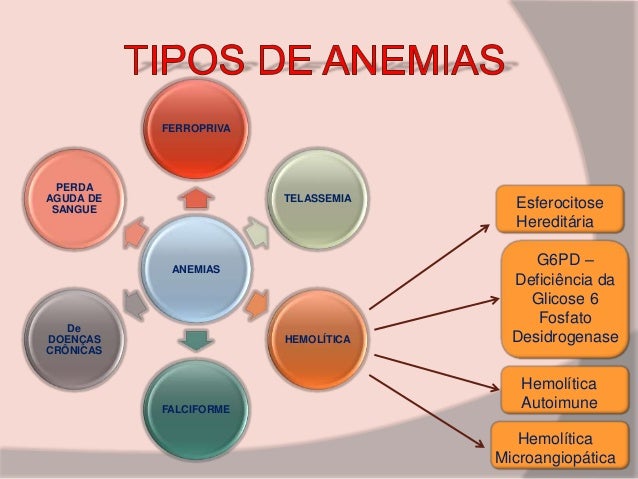
Anemia falciforme e outras hemoglobinopatias
Alterações estruturais na hemoglobina que prejudicam o transporte de oxigênio através das artérias e veias. Isso pode levar à oxigenação deficiente do organismo. Em geral, são classificadas em dois grupos: doença falciforme e talassemias.
As principais complicações clínicas da anemia falciforme são tratadas com as seguintes medidas profiláticas: antibióticos, suplementação de ácido fólico, nutrientes e rastreio de complicações.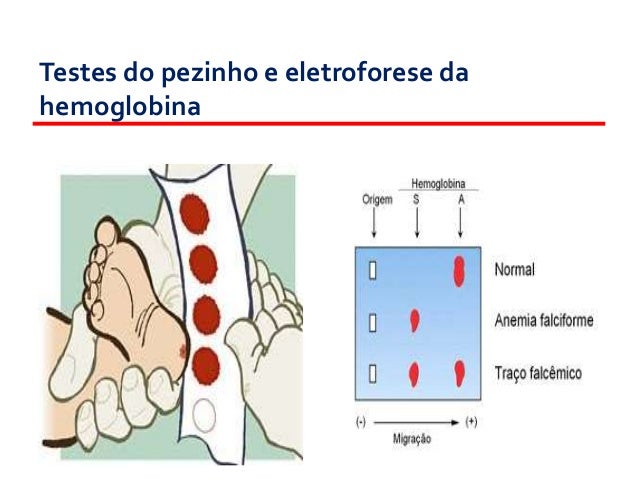
Os pacientes talassêmicos podem ser tratados através de um regime de transfusões, terapia quelante intensiva e esplenectomia, na tentativa de reduzir as necessidades de transfusão.
 Hiperplasia adrenal congênita
Hiperplasia adrenal congênita
As glândulas adrenais (suprarrenais) produzem diversos hormônios essenciais para o organismo. Para isso, elas dependem de enzimas específicas.
Quando uma destas enzimas está ausente, ocorre um desequilíbrio na produção dos hormônios levando a insuficiência adrenal e hiperandrogenismo.

Com maior frequência, o exame revela deficiência da enzima 21-hidroxilase.
Quando isso acontece, o cortisol é o hormônio que se torna deficiente e os hormônios andrógenos (masculinizantes) aumentam seus níveis. Em meninas, isso pode levar ao aparecimento de caracteres sexuais (pelos, aumento do clitóris etc.).
Em ambos os sexos, pode levar a uma perda acentuada de sal e óbito. Quando instituído correta e precocemente, o tratamento pode reverter esses quadros.

No caso da perda de sal, o tratamento requer a administração de hormônios mineralocorticoides com a máxima urgência. A suplementação de cortisona provoca a diminuição da síntese de hormônios androgênicos, relacionados à virilização.
Na forma perdedora de sal, a administração de mineralocorticoides deve ser continuamente monitorizada.
Fibrose cística
É uma doença genética, também conhecida como mucoviscidose, que causa mau funcionamento do transporte de cloro e sódio nas membranas celulares.
Esta alteração faz com que se produza um muco espesso nos brônquios e nos pulmões, facilitando infecções de repetição e causando problemas respiratórios e digestivos, entre outros.
Outra manifestação é o bloqueio dos ductos pancreáticos, causando problemas no sistema digestivo. Apesar de ainda não ter cura, diversas medidas terapêuticas têm melhorado a qualidade de vida e a sobrevida dos pacientes afetados.
Deficiência de biotinidase
A deficiência de biotinidase é uma doença metabólica hereditária na qual há um defeito no metabolismo da biotina.

Como consequência, ocorre uma depleção da biotina endógena devido a uma incapacidade do organismo fazer a sua reciclagem ou de usar a biotina ligada à proteína fornecida pela dieta. Assim, como a maioria dos erros inatos do metabolismo, esta doença apresenta uma herança autossômica recessiva.
Clinicamente, manifesta-se geralmente a partir da sétima semana de vida com distúrbios neurológicos e cutâneos, como crises epiléticas, hipotonia, microcefalia, atraso do desenvolvimento neuropsicomotor, alopécia (queda de cabelo) e dermatite eczematoide (“eczema”). Nos pacientes com diagnóstico tardio observam-se distúrbios visuais, auditivos assim como atraso motor e de linguagem.

O tratamento é simples e de baixo custo pois consiste na reposição oral de Biotina, na dose de 10 a 20 mg diariamente.
Desta forma, a deficiência de biotinidase preenche critérios para ser incluída na triagem neonatal (teste do pezinho): os pacientes afetados não mostram sinais clínicos neste período da vida, é uma doença com alta morbidade e que possui um tratamento efetivo.
Deficiência de glicose-6-fosfato desidrogenase (G6PD)
A G6PD é uma enzima presente em todas as células e tem como finalidade auxiliar na produção substâncias que as protegem de fatores oxidantes.
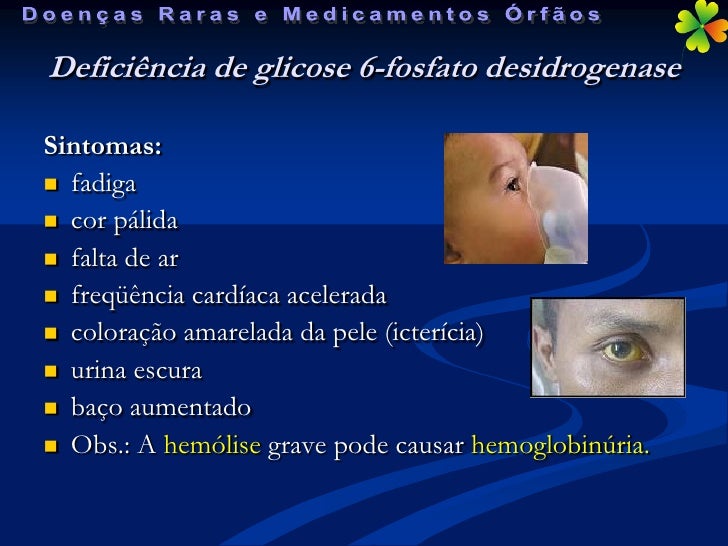
Ao contrário das outras células, os glóbulos vermelhos dependem exclusivamente da G6PD para esta finalidade. A deficiência de G6PD é uma doença genética associada ao cromossomo X e, ao contrário do que se esperaria, afeta igualmente indivíduos dos dois sexos.
A maior incidência ocorre em pessoas com ancestrais provenientes do Mediterrâneo, como Itália e Oriente Médio, da África Equatorial e de algumas regiões do Sudeste Asiático.
É reconhecida como a mais frequente das enzimopatias em algumas populações. Sua incidência no Brasil ainda não está estabelecida, mas estima-se que pode atingir até 7% da população.
 A doença não tem tratamento, mas seus sintomas podem ser evitados com medidas profiláticas que impeçam o uso de algumas drogas indutoras de hemólise e da ingestão do feijão de fava.
A doença não tem tratamento, mas seus sintomas podem ser evitados com medidas profiláticas que impeçam o uso de algumas drogas indutoras de hemólise e da ingestão do feijão de fava.
Os sintomas mais frequentes são icterícia neonatal e anemia hemolítica aguda. Em alguns casos a icterícia neonatal pode levar ao óbito ou a permanente dano neurológico. A anemia hemolítica pode ser induzida por um grande número de drogas, infecções ou pela ingestão de determinados alimentos.
Crianças com icterícia neonatal prolongada devem ser submetidas à fototerapia. Quando houver anemia, os pacientes podem necessitar de tratamento com oxigênio ou, em casos mais severos, transfusão sanguínea.
Fontes:
- http://www.soperj.org.br/novo/imageBank/TESTES-DE-TRIAGEM-NEONATAL.pdf
- http://www.sbteim.org.br/triagem-doencas-e-tratamentos.htm
- https://www.medicina.ufmg.br/nupad/seminario/biotinidase.htm